Publications
- A. Spentzos, S. May, A. Confer, M. Gau, P. Carroll, D. Goldberg, N. Tomson. "Investigating Metal–Metal Bond Polarization in a Heteroleptic Tris-ylide Diiron System". Inorg. Chem. (2023) (link)
- S. May, C. Climent, Z. Tao, S. A. Vinogradov, J. E. Subotnik. "Spin-orbit versus Hyperfine Coupling Mediated Intersystem Crossing in a Radical Pair". J. Phys. Chem. A., (2023) (link)
- S. L. De, S. May, K. Shah, M. Slawinski, S. Changrob, S. Xu, S. J. Barnes, P. Chootong, F. B. Ntumngia, J. H. Adams. "Variable immunogenicity of a vivax malaria blood-stage vaccine candidate". Vaccine, 39(19), 2668–2675. (2021) (link)
Research Experience
- Helpful overview in poster form
- Dr. John Adams
- Chen E, Salinas ND, Ntumngia FB, Adams JH, Tolia NH. (2015). Structural analysis of the synthetic Duffy Binding Protein (DBP) antigen DEKnull relevant for Plasmodium vivax malaria vaccine design. PLoS Negl Trop Dis. 9(3): e0003644.
- Chen E, Salinas ND, Huang Y, Ntumngia F, Plasencia MD, Gross ML, Adams JH, Tolia NH (2016). Broadly neutralizing epitopes in the Plasmodium vivax vaccine candidate Duffy Binding Protein. Proc Natl Acad Sci. 113(22):6277-82.
- Ntumngia, F.B*, Chootong, P*, VanBuskirk, KM*, Xainli, J, Cole-Tobian, JL, Campbell, CO, Fraser, TS, King, CL, Adams, JH (2010). Mapping epitopes of the Plasmodium vivax Duffy binding protein with naturally acquired inhibitory antibodies. Infection and Immunity. 78(3):1089-95.
- Ntumngia FB, Schloegel J, Barnes SJ, McHenry AM, Singh S, King CL, and Adams JH. (2012). Conserved and variant epitopes of Plasmodium vivax Duffy binding protein as targets of inhibitory monoclonal antibodies. Infection and Immunity. 80(3):1203-1208.
Research assistant in the Dr. Adams Lab (2018-2019)
I studied P. Vivax's DBPII protein under Dr. Sai Lata De at the University of South Florida. This was a formative foray into everything molecular biology, including mammilian cell cultures, transfection, transformation, PCR, flow cytometry, and basic immunologic assays like ELISAs and western blots. We published a paper on this work here
My project dealt with characterizing the effects of different variables on the immunigenicity of DBPII in both wild type and inbred mouse models. This research was inspired by previous work by Dr. Francis Ntumngia and others, and centers around DBPII's highly conserved nature and its ability to elicit high-titre inhibitory antibodies.
The reason for DBPII's highly conserved nature can be attributed to its function in the P. vivax parasite. P. vivax preferentially invades reticulocytes (young red blood cells) via binding between PvDBP and the reticulocyte's Duffy antigen receptor for chemokines (DARC). Region II of the DBP protein (DBPII) contains critical residues for heterotetramer formation when P. vivax invades the reticulocyte. Because of these critical residues, DBPII is a natural vaccine target.
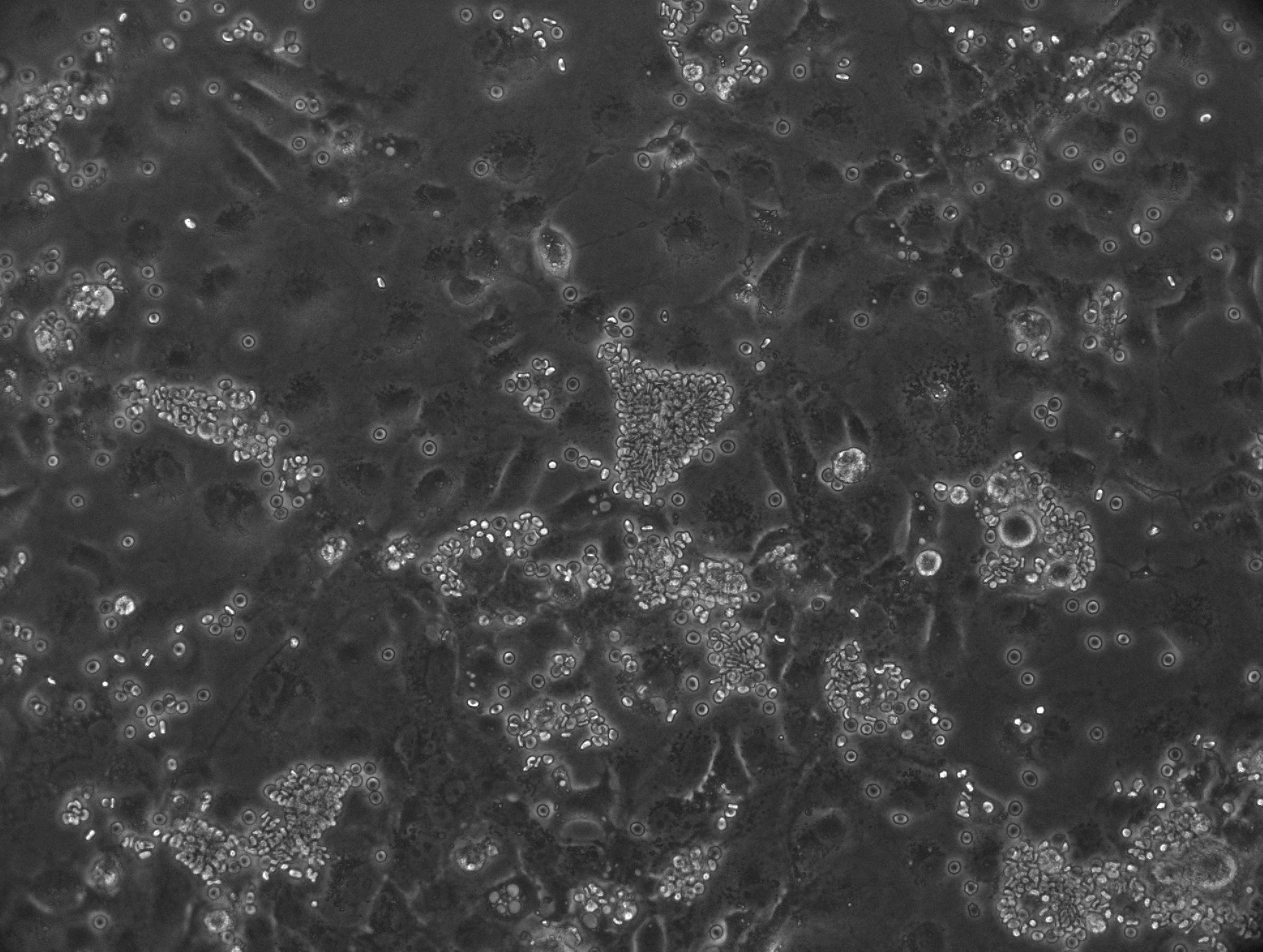
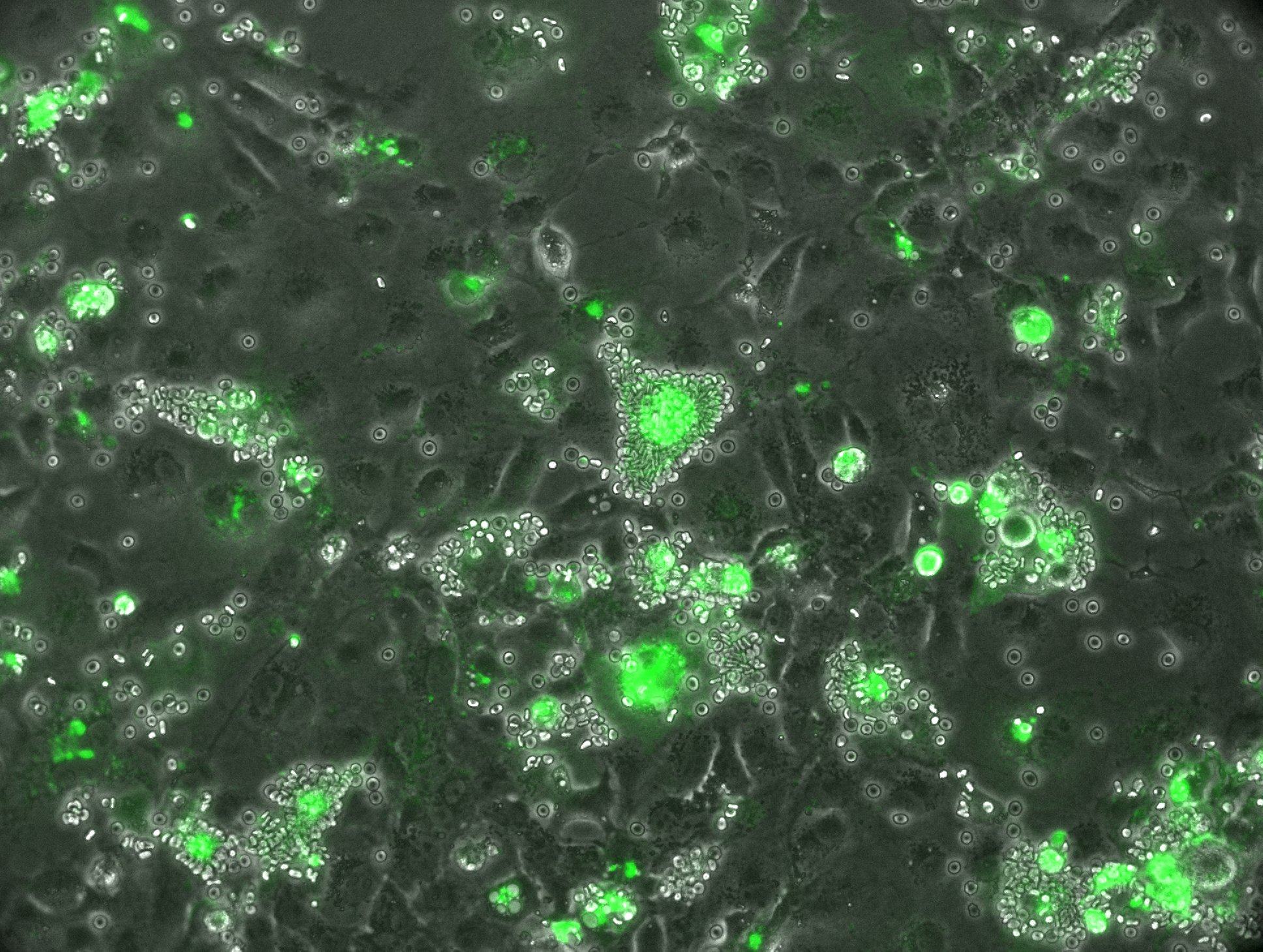
Inhibitory antibodies are a subset of antibodies that block a protein's active site, therefore inhibiting its function. This makes them especially desirable for vaccine design, as they can block critical antigens on a given pathogen. To evaluate the degree to which anti-DBPII mouse antibodies blocked DBPII-DARC heterotetramer formation, we used an in vitro inhibition assay. Cos7 cells were transiently transfected with DPII_GFP and were incubated in anti-DBPII mouse serum. The cells were then incubated in human erythrocytes to evaluate neutralization of the DBPII expressed on the surface of Cos7 cells. When functional inhibition of the mouse sera sample is lower, more "rosettes" (Cos7 cell covered in erythrocytes, shown below) will be observed.

Further reading and info
More photos


Undergraduate in the Dr. Tomson Lab (2020-2021)
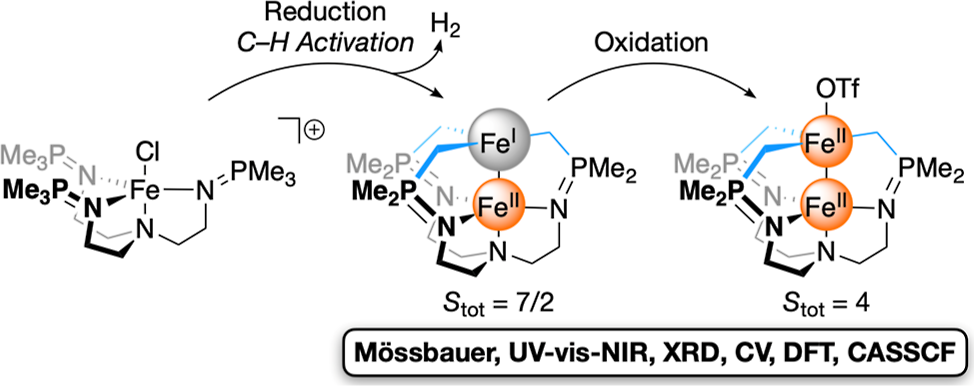
In the Dr. Tomson Lab, I helped to characterize a series of heteroleptic iron and diiron systems using an array of computational techniques in the Orca quantum chemistry package. Ground spin state and density were supported by DFT and CASSCF calculations. Additionally, experimental Mössbauer data was supported by carrying out linear fits with Mössbauer parameters of known similar molecules as a function of nuclear electron density. For synthetic and technical details, view the published work here.
Links and further reading
Undergraduate in Subotnik lab (2021-present)
Couplings mediating intersystem crossing in radical pairs
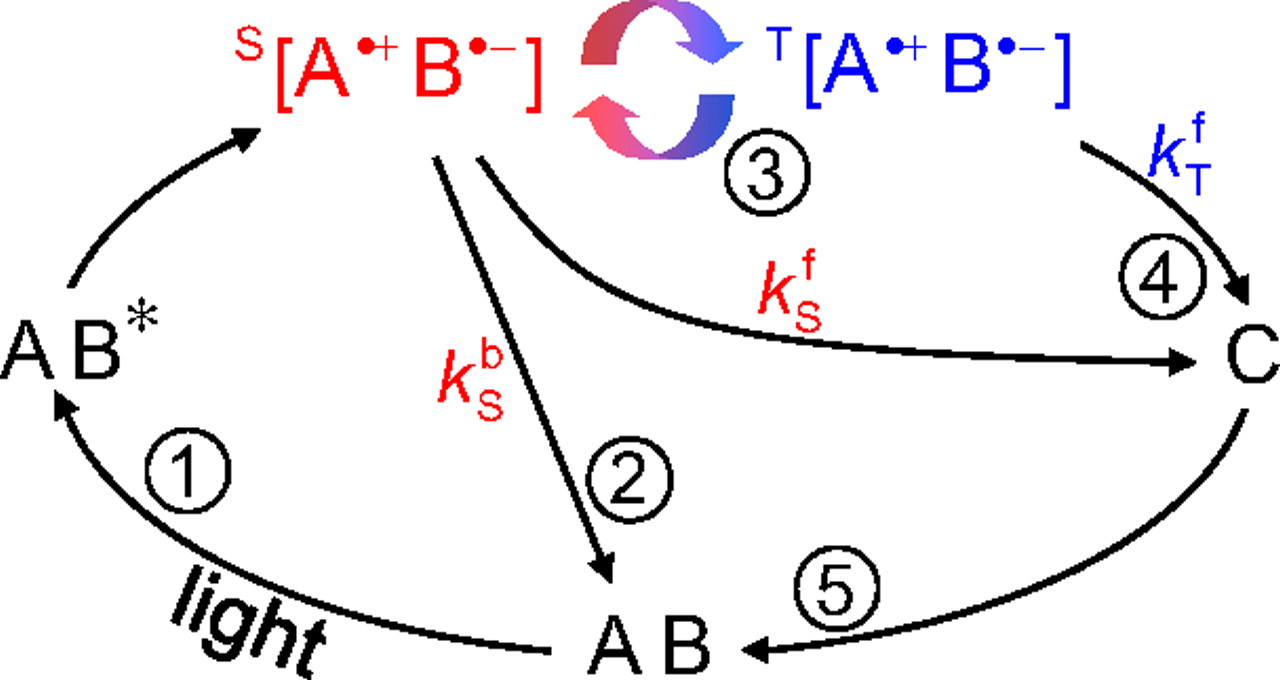
Radical pair (RP) systems consist of two correlated electrons localized on two molecules or molecular fragments. In the case of my research project, we were specifically interested in charge-transfer (CT) RP systems, in which there is a donor fragment and an acceptor fragment. Quantum mechanically, the two radicals are treated as a single coherent system which can be in the singlet state or one of 3 triplet states. These states are coupled together by two (main) mechanisms: spin-orbit coupling (SOC), in which the orbital angular momentum of an electron couples with the spin-angular momentum, and hyperfine coupling (HFC), in which the spin of an electron couples with the spin of nearby nuclei.
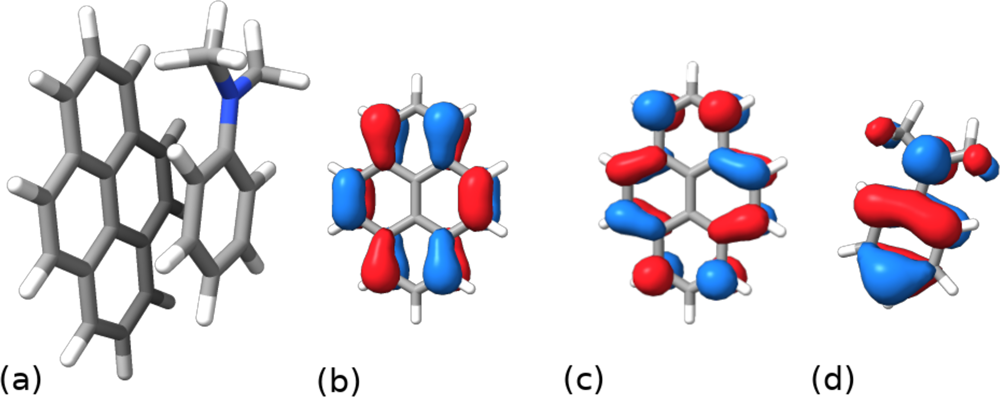
Bottom: Pyrene-dimethyaniline radical pair with HOMO/LUMO of Pyrene, and HOMO of dimethylaniline respectively. In the notation of the above figure, Pyrene is B and dimethylaniline is A.
In particular, we were interested in the relative strengths of SOC and HFC as a function of radical pair distance. To this end, I extended the Q-chem quantum chemistry program to calculate the coupling matrix elements between excited radical pair states. To test this code, we used a model radical pair Pyrene-dimethylaniline and calculated couplings at different donor-acceptor distances. We found that for this particular model, there is a small SOC-dominant regime beyond which the SOC goes to zero due to symmetry of the singlet-triplet states (since the exchange interaction goes to 0). For more info (and esoteric technical details), view the published work here. The hope is that this work provides a framework for designing Donor-Bridge-Acceptor or free radical pairs with tunable ISC rates.
My current work aims to extend this project to study other spin-related and EPR visible molecular characteristics.
Adding arbitrary relaxation to surface hopping
My first project in the Subotnik group focused on adding arbitrary electronic relaxation into a surface hopping model. It was partially successful, but is thus far unpublished. For a comprehensive review of surface hopping, see this review by Dr. Subotnik and Tully's original paper. The aim of the project was to model both non-adiabatic dynamics (dynamics in which the Born-Oppenheimer approximation breaks down and nuclear motion becomes coupled with electronic motion) and electron-hole photorelaxation in a single surface hopping framework to study small molecule charge transfer near metal surfaces. Two main difficulties arise in such a process.
The first is that an arbitrary relaxation term does not necessarily lend itself to a surface hopping rate. As seen in Tully's original paper, non-adiabatic hopping rates are calculated while relying on the fact that the quantum Liouville equation is formatted as a sum of commutators between operators and the density matrix (see eq. 11 in original paper). There is no reason that an arbitrary electronic relaxation (read: a damping term in the quantum liouville equation) should satisfy this commutator relation. The second is a less immediate problem of accomodating the high density of states (compared to an isolated molecular system) of a metal surface.